THE SAFETY
OF BRUKINSA
OVERALL INCIDENCE OF ADVERSE REACTIONS (ARs)1,2
| Adverse Reactions1,2 |
ARs in ≥10% of patients with WM (Cohort 1) | Pooled data: ARs in patients with hematologic malignancies |
||||
|---|---|---|---|---|---|---|
| BRUKINSA (n=101) | Ibrutinib (n=98) | BRUKINSA (N=1550)* | ||||
| All Grades (%) | Grades 3 or 4 (%) | All Grades (%) | Grades 3 or 4 (%) | All Grades (%) | Grade ≥3 (%) | |
| Upper respiratory tract infection | 44 | 0 | 40 | 2 | 39 | 2 |
| Pneumonia | 12 | 4 | 26 | 10 | 20 | 11 |
| Urinary tract infection | 11 | 0 | 13 | 2 | 13 | 2 |
| Diarrhea | 22 | 3 | 34 | 2 | 19 | 2 |
| Nausea | 18 | 0 | 13 | 1 | 11 | 0.2 |
| Constipation | 16 | 0 | 7 | 0 | 13 | 0.3 |
| Vomiting | 12 | 0 | 14 | 1 | 7 | 0.3 |
| Fatigue | 31 | 1 | 25 | 1 | 17 | 1 |
| Pyrexia | 16 | 4 | 13 | 2 | 10 | 0.8 |
| Edema peripheral | 12 | 0 | 20 | 0 | 4 | 0.2 |
| Bruising | 20 | 0 | 34 | 0 | 23 | 0.1 |
| Rash | 29 | 0 | 32 | 0 | 28 | 0.9 |
| Pruritus | 11 | 1 | 6 | 0 | 7 | 0.1 |
| Musculoskeletal pain | 45 | 9 | 39 | 1 | 30 | 2 |
| Muscle spasms | 10 | 0 | 28 | 1 | 5 | 0.1 |
| Headache | 18 | 1 | 14 | 1 | 11 | 0.4 |
| Dizziness | 13 | 1 | 12 | 0 | 11 | 0.3 |
| Cough | 16 | 0 | 18 | 0 | 19 | 0.1 |
| Dyspnea | 14 | 0 | 7 | 0 | 8 | 0.5 |
| Hemorrhage | 42 | 4 | 43 | 9 | 30 | 4 |
| Hypertension | 14 | 9 | 19 | 14 | 14 | 7 |
Safety in WM consistent with established BRUKINSA profile across B-cell malignancies1,2
The median follow-up time for Cohort 1 was 19.4 months.3
BRUKINSA had lower rates of:
Hypertension3
- Ibrutinib patients experienced nearly 2-fold higher incidence rate of hypertension on an exposure-adjusted basis (BRUKINSA vs ibrutinib: 0.7% vs 1.2%, respectively)
Major hemorrhage3
- Ibrutinib patients experienced nearly 2-fold higher incidence rate of major hemorrhage on an exposure-adjusted basis (BRUKINSA vs ibrutinib: 0.3% vs 0.6%, respectively)
INCIDENCE OF LABORATORY ABNORMALITIES
Select laboratory abnormalities† (≥20%) that worsened from baseline in Cohort 1
| Laboratory Abnormality1 | BRUKINSA‡ | Ibrutinib‡ | ||
|---|---|---|---|---|
| All Grades (%) | Grades 3 or 4 (%) | All Grades (%) | Grades 3 or 4 (%) | |
| Hematologic abnormalities | ||||
| Neutrophils decreased | 50 | 24 | 34 | 9 |
| Platelets decreased | 35 | 8 | 39 | 5 |
| Hemoglobin decreased | 20 | 7 | 20 | 7 |
| Chemistry abnormalities | ||||
| Bilirubin increased | 12 | 1.0 | 33 | 1.0 |
| Calcium decreased | 27 | 2.0 | 26 | 0 |
| Creatinine increased | 31 | 1.0 | 21 | 1.0 |
| Glucose increased§ | 45 | 2.3 | 33 | 2.3 |
| Potassium increased | 24 | 2.0 | 12 | 0 |
| Urate increased | 16 | 3.2 | 34 | 6 |
| Phosphate decreased | 20 | 3.1 | 18 | 0 |
INCIDENCE OF ATRIAL FIBRILLATION/FLUTTER
| Adverse Event3 |
All Grades n (%) |
Grade ≥3 n (%) |
||
|---|---|---|---|---|
| BRUKINSA (n=101) |
Ibrutinib (n=98) |
BRUKINSA (n=101) |
Ibrutinib (n=98) |
|
| Atrial fibrillation/flutter | 2 (2) | 15 (15) | 0 (0) | 4 (4) |
BRUKINSA HAD LOWER RATES OF:
Atrial fibrillation/flutter2,3
- Ibrutinib patients experienced nearly 10-fold higher incidence of atrial fibrillation/flutter on an exposure-adjusted basis (BRUKINSA vs ibrutinib: 0.1% vs 1.0%, respectively)
- No incidences of Grade ≥3 atrial fibrillation or flutter in patients who received BRUKINSA
LOW RATE OF ATRIAL FIBRILLATION/FLUTTER
AND HYPERTENSION WITH BRUKINSA
Initial analysis (19 months)3
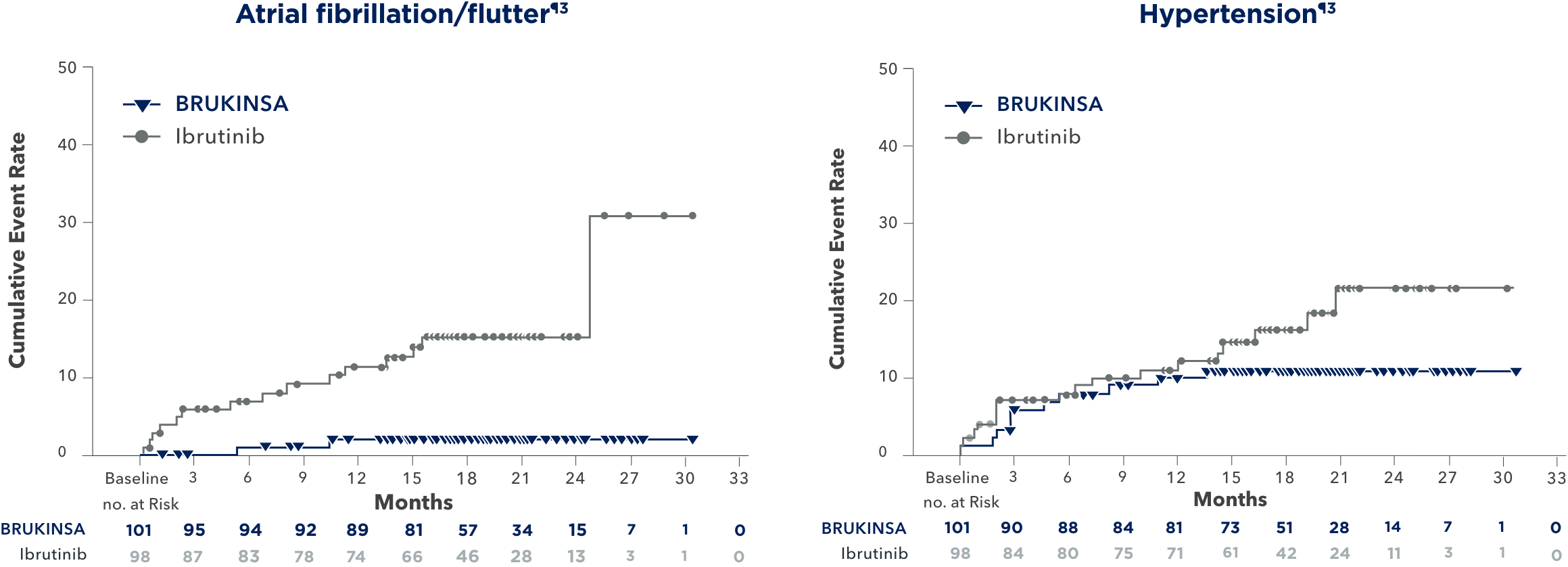
ADVERSE REACTIONS OF INTEREST AND THEIR PREVALENCE OVER TIME4
Long-term analysis (44 months)4

DOSE REDUCTIONS AND DISCONTINUATION RATE
DUE TO ADVERSE EVENTS (AEs) IN ASPEN (STUDY 302)3
Initial analysis (19 months)3
Dose reductions due to AEs
BRUKINSA
Cohort 1 (n=101)
of patients
Ibrutinib
(n=98)
of patients
Discontinuation rate due to AEs
BRUKINSA
Cohort 1 (n=101)
of patients
Ibrutinib
(n=98)
of patients
Median duration of treatment: 25 months2
In a ~4-year follow-up consistent with the primary analysis, fewer AEs leading to treatment discontinuation and dose reductions occurred with BRUKINSA4
Dr Anthony Nguyen discusses the safety profile of BRUKINSA vs ibrutinib in WM
References: 1. BRUKINSA. Package insert. BeiGene USA, Inc.; 2025. 2. Data on file. BeiGene USA, Inc. 3. Tam CS, Opat S, D’Sa S, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood. 2020;136(18):2038-2050. 4. Tam CS, Garcia-Sanz R, Opat S, et al. ASPEN: long-term follow-up results of a phase 3 randomized trial of zanubrutinib versus ibrutinib in patients with Waldenström macroglobulinemia. Poster presented at: American Society of Clinical Oncology (ASCO) 2022 Annual Meeting; June 3-7, 2022. Abstract 7521.